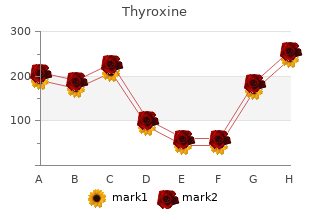
Pennsylvania State University at Altoona. V. Larson, MD: "Buy Thyroxine online - Quality Thyroxine no RX".
Pregnant women onpheny- toin should take folic acid each day to helpprevent neural tube defects buy cheap thyroxine 200 mcg online medicine used for adhd. Transient blood-clotting defects have been reportedinnew- borns whose mothers were taking this drug cheap thyroxine 100mcg otc symptoms influenza,butvitaminKgiven to mothers during the last month of pregnancy prevents this problem discount thyroxine 125mcg with mastercard medicine images. Phenytoin isexcretedinto breast milk in lowconcentrations cheap thyroxine 25mcg 911 treatment center, but it is considered safe to breast-feed full-term babies while taking this drug. The drug crosses the placenta and has beenuseful for controlling fetal supraventricular tachycardias. It isexcretedinto breast milk but has not been reported to cause problems in nursing infants. Propafenone should be avoidedduring pregnancybecause par- ticularly little information exists about its safety. Propafenone also isexcretedinto breast milk but has not been recognized to cause problemstonursing babies. Moricizine, like propafenone, has not been studiedinpregnant women and should be avoided. How- ever, reports suggest that beta blockers may be associatedwith low birth weights, neonatal bradycardiaand hypoglycemia. The most common antiarrhythmic application of beta blockers, in gen- eral, istocontrol the heart rate during atrial fibrillation. When controlling the ventricular response in atrial fibrillationduring pregnancy, attempts should be made first with digoxin and ve- rapamil, turning to beta blockers only if these are ineffective. Most beta blockers are excretedinto breast milk, but it is gener- ally considered safe to nurse full-terminfants while taking beta blockers. However, its impressive end-organ toxicity and its prolonged half-life mandate that itbe used only as a last resort during pregnancy. In addition to the array of “typical” amiodarone-related toxicities, risks specifically associ- atedwith pregnancy include premature labor, low birth weight, and neonatal hypothyroidism and hyperthyroidism. Amiodaroneap- pears in breast milk, and mothers taking this drug shouldnot breast- feed. Sotalol has not beenused widely or studied adequately during pregnancyand should be avoided. Itisexcretedinto breast milk, and its use during breast-feeding is not known to be safe. The drug does inhibit uterine contractions, which in fact has led to its use in inhibiting premature labor. Verapamil isexcretedinto breast milk but has noknown adverse effects onnursing babies. Itisexcretedinto breast milk and, ideally, should be avoidedinmothers who are breast-feeding. Therefore, this procedure should virtually never be performedduring pregnancy—again, with the exception of alife-threatening arrhythmia for which no other viable treatment option exists. Index acid-base disturbances, 13, 26, 28t reentrant arrhythmia, worsening acidosis, 47, 66 of, 118, 120–121 acute cardiac ischemia, 13 worsening of hemodynamics, 122 acute myocardial ischemia, 26, 75 afterpolarizations. See elimination/half-life from amiodarone, 94, 166 headaches and atrial fibrillation/atrial from adenosine, 109 flutter, 141t from dofetilide, 100 hypoglycemia from moricizine, 79 and beta blockers, 85, 166 from quinidine, 59 from disopyramide, 63 heart, electrical system and mexiletine, in newborn, anatomy, 4 (fig. The particular response to a drug by a patient is driven in one way or another by the concentration of that drug, and sometimes its metabolites, at the effect sites within the body. Accordingly, it is useful to partition the relationship between drug administration and response into two phases, a pharmacokinetic phase, which relates drug administration to concentrations within the body produced over time, and a pharmacodynamic phase, which relates response (desired and undesired) produced to concentration. In so doing, we can better understand why patients vary in their response to drugs, which includes genetics, age, disease, and the presence of other drugs. In other cases, the patient is suffering from several conditions, each of which is being treated with one or more drugs. Given this situation and the many potential sites for inter- action that exist within the body, it is not surprising that an interaction may occur between them, whereby either the pharmacokinetics or the pharmacodynamics of one drug is altered by another. More often than not, however, the interaction is of no clinical significance, because the response of most systems within the body is 1 2 Rowland graded, with the intensity of response varying continuously with the concen- tration of the compound producing it. Only when the magnitude of change in response is large enough will an interaction become of clinical significance, which in turn varies with the drug. For a drug with a narrow therapeutic window, only a small change in response may precipitate a clinically significant inter- action, whereas for a drug with a wide margin of safety, large changes in, say, its pharmacokinetics will have no clinical consequence. Also, it is well to keep in mind that some interactions are intentional, being designed for benefit, as often arises in combination therapy. Clearly, those of concern are the unintentional ones, which lead to either ineffective therapy through antagonism or lower concentrations of the affected drug or, more worryingly, excessive toxicity, which sometimes is so severe as to limit the use of the offending drug or, if it produces fatality, result in its removal from the market. This chapter lays down the conceptual framework for understanding the quantitative and temporal aspects of drug-drug interactions, hereafter called drug interactions for simplicity. Emphasis is placed primarily on the pharmacokinetic aspects, partly because pharmacokinetic interactions are the most common cause of undesirable and, to date, unpredictable interactions and also because most of this book is devoted almost exclusively to this aspect and indeed to one of its major components, drug metabolism. Some pharmacodynamic aspects are also covered, however, for there are many similarities between pharmacokinetic and pharmacodynamic interactions at the molecular level and because ultimately one has to place a pharmacokinetic interaction into a pharmacodynamic perspective to appreciate the likely therapeutic impact (1–5). Absorption, which applies to all sites of administration other than direct injection into the bloodstream, comprises all processes between drug administration and appearance in circu- lating blood. Disposition comprises both the distribution of a drug into tissues within the body and its elimination, itself divided into metabolism and excretion of unchanged drug. Disposition is characterized independently following intra- venous administration, when absorption is not involved. Increasingly, aspects of potential drug interactions are being studied in vitro not only with the aim of providing a mechanistic understanding but also with the hope that the findings can be used to predict quantitatively events in vivo, and thereby avoid or limit undesired clinical interactions. To achieve this aim, we need a holistic approach whereby individual processes are nested within a whole body frame—that is, constructs (models) that allow us to explore the impact, for example, of inhibition or induction of a particular metabolic pathway on, say, the concentration–time profile of a drug in the circulating plasma or blood, which delivers the drug to all parts of the Introducing Pharmacokinetic and Pharmacodynamic Concepts 3 Figure 1 Schematic representation of processes comprising the pharmacokinetics of a compound. Absorption comprises all events between drug administration and appearance at the site of measurement. Distribution is the reversible transfer of the drug from and to other parts of the body. Elimination is the irreversible loss of the drug either as unchanged compound (excretion) or by metabolism. Disposition is the movement of the drug out of blood by distribution and elimination. This approach also allows us to better interpret the underlying events occurringinvivofollowingadruginteraction. To appreciate this last statement, consider the events shown in Figures 2 and 3 and the corresponding summary data given in Table 1. As can be seen, these clinical studies show clear evidence of an interaction, with both actually involving the same mechanism, enzyme induction, but the effect is clearly expressed in different ways. To understand why this is so, we need to deal first with the intravenous data and then with the oral data—that is, to separate disposition from absorption. For many purposes, because distribution is often much faster than elimi- nation, as a first approximation the body can be viewed as a single compartment, of volume V, into which drugs enter and leave. This is an apparent volume whose value varies widely among drugs, owing to different distribution patterns within the body. The larger the volume, the lower the plasma concentration for a given amount in the body.
Another advantage is that the probe can be anchored in place with dental cement and experiments carried out later cheap thyroxine 75 mcg on line symptoms quivering lips, in conscious freely moving animals once they have recovered from the anaesthetic purchase thyroxine 200 mcg otc medicine 54 543. Indeed cheap 125 mcg thyroxine amex medicine that makes you throw up, comparison of results from studies carried out on both anaesthetised and freely moving subjects has revealed drug interactions with anaesthetics that can affect transmitter release: anaesthetic-induced changes in the regulation of noradrenaline release by a2-adrenoceptors is a case in point buy generic thyroxine 200mcg online symptoms ear infection. The length of membrane below the probe support can be altered (1±10 mm)to suit the size of the animal and the brain area being studied. Flow rates are normally below 2 ml/min or repeated studies on the same animals but this requires a slight modification of the technique. Unfortunately, for a variety of reasons, each microdialysis probe can be used for only a few hours and so it has to be replaced each day. However, the presence of the guide cannula makes this a relatively straightforward process that requires only light sedation of the animal. A further advantage of microdialysis is that, unlike the push±pull cannula or the cortical cup, the perfusion medium does not come into direct contact with the tissue being studied. This reduces damage caused by turbulence as well as enzymic degradation of the transmitter. For instance, acetylcholine, but not cholinesterase, will penetrate the probe membrane. Finally, because solutes will pass out of the probe, as well as into it, the probe can also be used for infusing ions (Fig. This avoids many of the problems that arise when trying to determine the synaptic actions of drugs when these are administered systemically. The graph shows efflux of noradrenaline in the frontal cortex of anaesthetised rats. Increasing the concentration of K in the medium infused via the probe increases noradrenaline efflux whereas removing Ca2 reduces it is needed for analysis: i. In any case, the efficiency of the probe membrane limits the net influx (or efflux)of solutes to about 10± 20% of the theoretical maximum. It should also be borne in mind that the microdialysis probe is not sampling the transmitter in the synapse: only that transmitter which escapes metabolism in, or reuptake from, synapses will migrate towards the probe. In the drug-free state, any change in the transmitter concentration in the dialysis samples is usually assumed to indicate a change in its rate of release from nerve terminals; this is supported by the spontaneous efflux of transmitters being Ca2-dependent and K- sensitive (Fig. However, efflux does not always reflect release rate, especially if experimental interventions (e. Voltammetry This can be carried out in vitro (in brain slices, cultured cell preparations)or in vivo and involves penetrating the experimental tissue with a carbon-fibre electrode of 5±30 mmin diameter (Fig. This serves as an oxidising electrode and the Faradaic current generated by the oxidation of solutes on the surface of the electrode is proportional to their concentration. Obviously, only neurotransmitters which can be oxidised can be measured in this way so the technique is mainly limited to the study of monoamines and their metabolites. Changes in the concentration of transmitters are monitored by rapid cycles of voltage scans (e. Since a complete scan takes only about 20 ms, the time resolution with voltammetry is much better than with microdialysis and is suitable for studying rapid, transient changes in transmitter release. One difficulty with this method is that all oxidisable solutes in the external medium will be incorporated into the voltammogram and interfering peaks can be a problem. In fact, the concentration of monoamine metabolites and oxidisable solutes can be considerably greater than those of the parent amines which can be difficult to distin- guish as a result. Ascorbic acid and uric acid are particularly problematic in this respect, although recent work suggests that an increase in the concentration of extracellular ascorbic acid could be a marker for the early phase of cerebral ischaemia. In general, voltammetry is most useful for measuring rapid (subsecond)changes in monoamine release. Under these circumstances, slower changes in the metabolites and other compounds do not interfere. The trace is a plot of the oxidation peak height against time, calibrated for dopamine. One of the earliest biosensors was the dorsal wall muscle of the leech which contracts in the presence of nM concentrations of acetylcholine. Others are the bioluminescent proteins, such as aequorin, which fluoresce in the presence of Ca2. Within a reasonable range, the fluorescence intensity is pro- portional to the cation concentration and so it can be used to monitor the increase in the intracellular concentration of Ca2 during excitation of nerve terminals. More recently, biosensors have been developed which comprise electrodes coated with glucose oxidase or lactate oxidase. The activity of these enzymes generates a current that can be used to quantify the concentration of glucose and lactate on the surface of the electrode. This work is playing an important part in research on brain metabolism during neuronal activity. Two separate lines of research led to the proposal that transmitter released in response to neuronal excitation is derived from a vesicle-bound pool rather than from the neuronal cytoplasm. Using differential centrifugation, these vesicles were soon identified as the major storage sites for neurotransmitters. The second was electrophysiological evidence that the effect of neuronal release of acetylcholine on the postsynaptic membrane potential at the neuromuscular junction was quantal in nature, suggesting that this transmitter, at least, was released in discrete packets. Early neurochemical investigations of the source of released transmitter measured noradrenaline release from chromaffin granules in the adrenal medulla. Chromaffin granules are considerably larger (250 nm diameter)than the storage vesicles in noradrenergic nerve terminals (40±100 nm)and so their experimental use avoided the constraint imposed by the low sensitivity of early assay techniques (see Winkler 1993). Yet, like noradrenergic neurons, the adrenal medulla is derived from the developing neural crest and noradrenaline release is activated by stimulation of preganglionic cholinergic neurons. Chromaffin granules therefore provide a useful model for pro- cesses involved in the storage and release of noradrenaline from neurons. Subsequent refinements of assays for noradrenaline enabled studies of noradrenaline release to be extended to stimulated sympathetic nerve/end-organ preparations. These experiments confirmed that noradrenaline was released from vesicle-bound packets of transmitter contained within the terminal vesicles. Experiments of this kind have provided a great deal of evidence in favour of exocytotic release of vesicular noradrenaline. For example, by administering reserpine (which causes noradrenaline to leak out of the vesicles into the cytoplasm)together with an inhibitor of the enzyme monoamine oxidase (which will prevent metabolism of cytoplasmic noradrenaline), it is possible to redistribute the noradrenaline stored within nerve terminals because it leaks from the vesicles but is preserved within the neuronal cytoplasm. Under these conditions, the total amount of transmitter in the terminals is unchanged but impulse-evoked release rapidly diminishes. Different evidence, mainly based on histological studies, suggested that acetylcholine is also released by vesicular exocytosis. It is then possible to fracture axolemma membranes in a way that separates their lipid bilayer. Electron microscopy reveals numerous pits in the membranes which are thought to reflect the vesicle/axolemma fusion pore of vesicles in the process of exocytosis. Subsequent studies, combining immunocytochemistry with electron microscopy, showed that proteins in the membranes of vesicles become incorporated into the axolemma during transmitter release. Furthermore, when neurons are stimulated in a medium containing an electron-dense marker, that does not penetrate the neuronal membrane, the marker later appears in vesicles inside the nerve terminals (Basbaum and Heuser 1979). This suggests that such markers are incorporated into the vesicles when they come into contact with the extracellular fluid during exocytosis.
The stereoselective metabolism of fluoxetine in poor and extensive metabolisers of sparteine buy cheap thyroxine 50 mcg line treatment skin cancer. The role of cytochrome P-450D6 in the metabolism of paroxetine by human liver microsomes generic 75mcg thyroxine mastercard symptoms 7dpiui. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabo- lism discount 200 mcg thyroxine otc symptoms 8dpo. Identification of three cyto- chrome P-450 isozymes involved in N-demethylation of citalopram enantiomers in human liver microsomes buy generic thyroxine 100mcg line medications valium. A fatal case of serotonin syndrome after combined moclobemide- citalopram intoxication. Postmortem forensic toxicology of selective serotonin reuptake inhibitors: a review of pharmacology and report of 168 cases. Plasma concentra- tions of risperidone and 9-hydroxyrisperidone during combined treatment with paroxetine. Fluvoxamine and fluoxetine do not interact in the same way with the metabolism of the enantiomers of methadone. Selective serotonin reuptake inhibitors and tricyclic antidepressants in combina- tion. Rifampin induced selective serotonin reuptake inhibitor with- drawal syndrome in a patient treated with sertraline. Effect of selective serotonin reuptake inhib- itors on the oxidative metabolism of propafenone: in vitro studies using human liver micro- somes. Inhibition of phenytoin hydroxylation in human liver microsomes by several selective serotonin reuptake inhibitors. Lack of effect of citalopram on the steady state pharmacokinetics of carbamazepine in healthy male subjects. Paroxetine and amitrip- tyline augmentation of lithium in the treatment of major depression: a double-blind study. Tolerability of com- bined treatment with lithium and paroxetine in patients with bipolar disorder and depres- sion. Paroxetine affects metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Dose-response evaluation of the inter- action between sertraline and alprazolam in vivo. Ayahuasca preparations and serotonin reuptake inhibitors: a potential combination for severe adverse interactions. Treatment of depression with associated anxiety: compari- sons of tricyclic and selective serotonin reuptake inhibitors. Adverse effects associated with selective serotonin reuptake inhibitors and tricyclic antidepressants: a meta-analysis. Seizure risk associated with psychotropic drugs: clinical and pharmacoki- netic considerations. Is therapeutic drug monitoring a case for optimizing clinical outcome and avoiding interactions of the selective serotonin reuptake inhibitors? Therapeutic drug monitoring of selective serotonin reuptake inhibitors influences clinical dosing strategies and reduces drug costs in depressed elderly patients. A double blind, placebo-controlled study of citalopram with and without lithium in the treatment of therapy- resistant depressive patients: a clinical, pharmacokinetic, and pharmacogenetic investiga- tion. Traditional antipsychotics are thought to act by exerting effects principally on the dopamine neurotransmitter system (1). The traditional antipsychotics became known to many as neuroleptics based on their frequent effects of substantially slowing movement (1). Atypical antipsycho- tics, designed in laboratories to provide psychotic symptom relief without movement problems, affect other neurotransmitter systems (2), and present other potential concerns. Interactions involving antipsychotics (a) make side effects of the antipsychotics more pronounced, (b) render the antipsychotics less effective, and (c) affect the metab- olism of other medicines, and prolong their effects and side effects Both older and more recently developed varieties of antipsychotics are known for their manifold side effects on numerous organ systems. Interactions have forensic sig- nificance when efficacy and/or side effects are heightened by the coprescription of med- icines that affect antipsychotic metabolism. Drug interactions involving antipsychotics warrant particular scrutiny in the elderly, the brain-damaged, those on other psychotropics, and those with a history of special sensitivity to antipsychotics. Given the severe conditions for which antipsychotic prescribing is reserved, inter- actions also have forensic relevance when an antipsychotic is no longer effective because of the medicines prescribed along with it. In these cases, the greatest forensic significance of the drug interaction is the relapse of the root illness, rather than drug side effects. Before we explore how these interactions manifest themselves in criminal and civil case scenarios, an appreciation for the neurochemistry involved is necessary. Messages pass through the nervous system, from cell to cell, by these chemical neurotransmitters (3). Psychosis and other psychiatric maladies occur when the delicate equilibrium of each of these micro- scopic neurochemical transmitters is disrupted. The chemical imbalance causes chain reactions that result in the development of symptoms or outwardly visible behaviors. Antipsychotics impact a number of neurotransmitters and regulatory systems in the body. Like other psychotropics, antipsychotics exert their effects on receptors of these neurotransmitters, receptors that normally catch and relay the transmitting neuro- chemical that has been released by the nerve cell nearby. In addition to directly blocking dopamine transmission at D2 receptors, anti- psychotics have antihistaminic and antiadrenergic effects (4). All traditional antipsy- chotics, particularly those that are classified as low potency, have anticholinergic effects focusing on the muscarinic class of receptors (5). The effects of such neurotransmitter blockade depend not only on the neurotransmitter, but where in the brain that neuro- transmitter is active, and what role in human functioning it plays. Atypical antipsychotic drugs earn their name, in part, because they do not cause effects on movement in the way traditional antipsychotic drugs do. Whereas each of the atypical antipsychotics impacts a distinct profile of neurotransmitters, all of the atypical class block dopamine D2 receptors, as well as serotonin 2A receptors (6). Dopamine: Benefits and Movement Problems Caused by Its Blockade Dopamine has been the foundation of antipsychotic treatment. Traditional antipsy- chotics’ influence on the different centers of dopamine activity has directly and indirectly accounted for side effects of forensic significance. Sagittal section of human brain showing the dopaminergic pathways involved in the actions of antipsychotic drugs. Psychotic illnesses and certain drug intoxications, such as cocaine and amphet- amines, arise from altered dopamine transmission. Traditional antipsychotics decrease or eliminate psychotic symptoms like hallucinations and delusions, and organize con- fused thinking, regardless of their origin. These medicines block dopamine transmis- sion at D2 receptors in the mesolimbic nerve pathways that lead to the nucleus accumbens in the limbic system of the brain (7). Therefore, regrettably, dopamine blocking in other areas of the brain results in unwanted conse- quences as well. Parkinsonism, dystonia, akathisia, tardive dyskinesia, and tardive dystonia stem, through a variety of mechanisms, from the capacity of antipsychotics to block dopa- mine transmission in the brain (8). Dopamine activity in the brain substantia nigra is necessary for unrestricted movement.