

Sacred Heart University. O. Campa, MD: "Order cheap Panmycin online no RX - Cheap online Panmycin OTC".
Sometimes cheap panmycin 500 mg otc antibiotic eye drops for pink eye, an absorption-related drug interaction can be avoided by administering the drugs at least 2 hours apart generic panmycin 250 mg line antibiotic probiotic. Bound and determined After a drug is absorbed panmycin 250 mg mastercard infection zit, the blood distributes it throughout the body as a free drug or one that’s bound to plasma protein order 500mg panmycin overnight delivery antibiotic treatment for strep throat. When two drugs are given together, they can compete for protein-binding sites, leading to an increase in the effects of one drug as that drug is displaced from the protein and becomes a free, unbound drug. Toxic waste Toxic drug levels can occur when a drug’s metabolism and excre- tion are inhibited by another drug. Some drugs stimulate enzyme production, increasing metabol- ic rates and the demand for vitamins that are enzyme cofactors (which must unite with the enzyme in order for the enzyme to function). For instance, when food that contains Vitamin K (such as green, leafy vegeta- bles) is eaten by a person taking warfarin, the drug’s anticoagula- tion properties are decreased and blood clots may form. Grapefruit can inhibit the metabolism of certain medications, resulting in toxic blood levels; examples include fexofenadine, albendazole, and atorvastatin. Because of all the interactions food can have with drug metabolism, being aware of drug interactions is essential. Adverse drug reactions A drug’s desired effect is called the expected therapeutic re- sponse. An adverse drug reaction (also called a side effect or ad- verse effect), on the other hand, is a harmful, undesirable re- sponse. Adverse drug reactions can range from mild ones that dis- appear when the drug is discontinued to debilitating diseases that become chronic. Adverse reactions can appear shortly after start- ing a new medication but may become less severe with time. Dosage dilemma Adverse drug reactions can be classified as dose-related or patient sensitivity–related. Most adverse drug reactions result from the known pharmacologic effects of a drug and are typically dose- related. Dose-related reactions include: • secondary effects • hypersusceptibility • overdose • iatrogenic effects. For example, morphine used for pain control can lead to two extreme sensitivity to undesirable secondary effects: constipation and respiratory de- a drug. Diphenhydramine used as an antihistamine produces se- dation as a secondary effect and is sometimes used as a sleep aid. Enhanced action A patient can be hypersusceptible to the pharmacologic actions of a drug. Such a patient experiences an excessive therapeutic re- sponse or secondary effects even when given the usual therapeu- tic dose. Hypersusceptibility typically results from altered pharmacoki- netics (absorption, metabolism, and excretion), which leads to higher-than-expected blood concentration levels. Increased recep- tor sensitivity also can increase the patient’s response to therapeu- tic or adverse effects. A toxic drug reaction can occur when an excessive dose is taken, either intentionally or by accident. The result is an exaggerated re- sponse to the drug that can lead to transient changes or more seri- ous reactions, such as respiratory depression, cardiovascular col- lapse, and even death. To avoid toxic reactions, chronically ill or elderly patients often receive lower drug doses. Iatrogenic issues Some adverse drug reactions, known as iatrogenic effects, can mimic pathologic disorders. Other examples of iatrogenic ef- fects include induced asthma with propranolol, induced nephritis with methicillin, and induced deafness with gentamicin. You’re so sensitive Patient sensitivity–related adverse reactions aren’t as common as dose-related reactions. Sensitivity-related reactions result from a patient’s unusual and extreme sensitivity to a drug. These adverse reactions arise from a unique tissue response rather than from an exaggerated pharmacologic action. Extreme patient sensitivity can occur as a drug allergy or an idiosyncratic response. Previous ex- posure to the drug or to one with similar chemical characteristics For an allergic sensitizes the patient’s immune system, and subsequent exposure reaction to occur, the patient must have causes an allergic reaction (hypersensitivity). The allergic reaction can vary in intensity from an immediate, life-threatening anaphylactic reaction with circulatory col- lapse and swelling of the larynx and bronchioles to a mild reaction with a rash and itching. Idiosyncratic response Some sensitivity-related adverse reactions don’t result from pharmacologic properties of a drug or from an allergy but are specific to the individual patient. While teaching a patient about drug therapy for diabetes, you review the absorption, distribution, metabolism, and excretion of insulin and oral antidiabetic agents. Pharmacokinetics discusses the movement of drugs through the body and involves absorption, distribution, metabo- lism, and excretion. Which type of drug therapy is used for a patient who has a chronic condition that can’t be cured? Maintenance therapy seeks to maintain a certain lev- el of health in patients who have chronic conditions. Pharmacodynamics studies the mechanisms of ac- tion of drugs and seeks to understand how drugs work in the body. Sometimes food enhances absorption—so grab a quick snack and come back for a review. Cholinergic drugs enhance the action of acetylcholine, stimulating the parasympathetic nervous system. Cholinergic drugs Cholinergic drugs promote the action of the neurotransmitter acetylcholine. These drugs are also called parasympathomimetic drugs because they produce effects that imitate parasympathetic nerve stimulation. Mimickers and inhibitors There are two major classes of cholinergic drugs: Cholinergic agonists mimic the action of the neurotransmit- ter acetylcholine. Anticholinesterase drugs work by inhibiting the destruction of acetylcholine at the cholinergic receptor sites. How cholinergic drugs work Cholinergic drugs fall into one of two major classes: cholinergic agonists and anticholinesterase drugs. Cholinergic agonists Anticholinesterase drugs When a neuron in the parasympathetic nervous system is stim- After acetylcholine stimulates the cholinergic receptor, it’s de- ulated, the neurotransmitter acetylcholine is released. Anticholinester- choline crosses the synapse and interacts with receptors in an ase drugs inhibit acetylcholinesterase. Cholinergic agonists stimulate cholinergic re- line isn’t broken down and begins to accumulate, leading to ceptors, mimicking the action of acetylcholine. Pharmacokinetics (how drugs circulate) The action and metabolism of cholinergic agonists vary widely and depend on the affinity of the individual drug for muscarinic or nicotinic receptors.
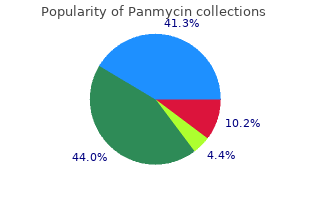
Syndromes
- Major mood swings
- General discomfort (malaise)
- Most people have no symptoms
- In some places you may be able to take information and medicines to your sexual partner yourself.
- Bronchoscopy with biopsy
- Maintain pressure until the bleeding stops. When it has stopped, tightly wrap the wound dressing with adhesive tape or a piece of clean clothing. Place a cold pack over the dressing. Do not peek to see if the bleeding has stopped.

However order 500mg panmycin with visa bacteria zinc, it is useful when comparing the effect of disease 250mg panmycin otc antibiotic no alcohol, altered physiologic state quality panmycin 250mg virus - zippy, or drug-drug interaction on the pharmacokinetics of a specific drug cheap 500mg panmycin amex antibiotics for uti in elderly. This parameter is not affected by changes in drug elimination or clearance, making it a useful tool in assessing the effect disease, altered physiologic state, or drug-drug interaction may have on the volume of distribution of a drug. Vss was calculated previously but was only applicable to a drug fitting a two-compartment model. Formation clearance is analogous to systemic and renal clearance of a drug and refers to the formation of metabolites in the course of drug elimination. The following equations are used to calculate the formation clearance of a drug: ClP→m1 = Fm1Clt where: ClP→m1 = fractional clearance of the parent drug (P) to form metabolite 1 (m1), Fm1 = fraction of metabolite m1 formed from a single dose of the parent drug, and Cl = total body clearance. For example, if a drug is metabolized by three separate enzyme systems, each producing a unique metabolite, what effect would the addition of a known hepatic enzyme inducer have on the individual metabolic pathways? To simplify this example, we will assume that systemic clearance equals hepatic clearance, these three metabolic pathways account for 100% of the hepatic clearance of the drug, the metabolite is rapidly secreted unchanged in the urine, and the dose is equal to 100 mg. Table 11-1 shows the effect of an enzyme inducer on each metabolic pathway portrayed in Figure 11-8 as shown by changes in the percentage of drug dose excreted in the urine for each metabolite and formation clearance. As Table 11-1 shows, the administration of an enzyme inducer substantially increased the systemic clearance of this drug, from 25 to 75 mL/minute. However, the change in the percentage of the dose excreted as a specific metabolite does not exactly reflect the change in formation clearance values. The percentage of dose excreted in the urine for m1 was reduced threefold, but no change in the formation clearance was observed. This means that the enzyme inducer had no effect on the enzyme responsible for producing m1. Finally, the percentage of dose excreted in urine for m3 was unchanged despite a threefold increase in its formation clearance. Because the formation clearance of a drug to a metabolite reflects more accurately the activity of that specific enzyme, the data would suggest that the enzyme(s) responsible for the formation of m2 and m3 are significantly increased by the enzyme inducer, whereas the enzyme(s) responsible for the formation of m1 was unaffected. The preceding example demonstrates the value of formation clearance versus the more traditional approach of calculating the percentage of a drug dose excreted as a specific metabolite. Metabolic pathways for a parent drug, where m1 = metabolite 1, m1,u = amount of m1 excreted in the urine, m2 = metabolite 2, m2,u = amount of m2 excreted in the urine, and m3 = metabolite 3, m3,u = amount of m3 excreted in the urine. Absorption of therapeutic drugs by barrier gels in serum separator blood collection devices. With dysfunction of the major organs of drug elimination (kidneys and liver), drug clearance, volume of distribution, and drug plasma protein binding may be affected. For drugs that distribute primarily in extracellular fluid, a dose for an obese person should be calculated using total body weight. The fluid portion of a sample of whole blood allowed to clot for 30 minutes before centrifugation is called: A. Indicate Yes or No for questions 11-7 through 11-10: Yes = the accuracy of the drug concentrations is of concern and should be redrawn. A gentamicin concentration from a sample stored at controlled room temperature and assayed 24 hours after it was collected from a patient receiving both ampicillin and gentamicin. A plasma tobramycin concentration from a sample stored at controlled room temperature and assayed 24 hours after it was collected from a patient receiving both tobramycin and ceftazidime. A plasma gentamicin concentration from a sample stored in a freezer until assayed 12 hours after it was collected from a patient receiving both ampicillin and gentamicin. A plasma gentamicin concentration from a sample assayed immediately after it was collected from a patient receiving both piperacillin and gentamicin. Which statement(s) is/are false about the calculation of formation clearance (ClP→mX)? The proportion of the body that is water is greatest in the neonate and lowest in the elderly. The proportion of fat tissue that is extracellular fluid is less than in lean tissue, but the drug will still distribute somewhat in the adipose extracellular fluid. Serous fluid is a natural body fluid and is not centrifuged to remove cellular components. Serous fluid is a natural body fluid and is not centrifuged to remove cellular components. Assay cross-reactivity does not involve assay measurement of inactivated products that result from some physiochemical process. Write a pharmacy protocol to ensure proper serum drug concentration collection and assay. Try to get a package insert from your laboratory on any therapeutically monitored drug. Specifically, how are the issues of assay sensitivity, specificity, and cross- reactivity noted? The relative bioavailabilities of two dosage forms (a sustained-release tablet and an oral solution) of oral morphine sulfate are being compared. The following plasma drug concentrations were obtained after 30 mg of each was administered: Before proceeding to the questions below, on graph paper (linear), plot the plasma drug concentration versus time data for the two formulations. What are the peak plasma drug concentrations for the oral tablet and oral suspension, respectively? What is the absorption rate constant (Ka) of this formulation (using the method of residuals)? First, the data points at 4, 8, and 12 hours are on the straight-line terminal portion of the plot and therefore are not used to calculate the residual line. Next, the terminal, straight-line portion of the graph is back-extrapolated to the y-axis. For each time at which a concentration was actually determined, the concentration corresponding to the back-extrapolated line is noted (extrapolated concentration). The residual is the remainder of the actual concentration subtracted from the extrapolated concentration. The Ka is the negative slope of the natural log of the residual concentration versus time curve. We can choose any two residual points to determine the slope, but it is usually best to select the points most widely separated by time. Aminoglycosides In this lesson, Cases 1-5 focus on the safe and appropriate dosing of aminoglycosides. Individualization of aminoglycoside dosing regimens is important to optimize efficacy while minimizing potential toxicity. Cases 1-4 outline traditional dosing methods of individualized dosing, and Case 5 focuses on the extended interval administration of aminoglycosides. Because the currently available intravenous aminoglycosides (gentamicin, tobramycin, and amikacin) exhibit similar pharmacokinetics, case discussions of one aminoglycoside can be extrapolated to any other. Although amikacin has the same pharmacokinetic profile as other aminoglycosides, it requires doses and target concentrations approximately four times as high as the other aminoglycosides.
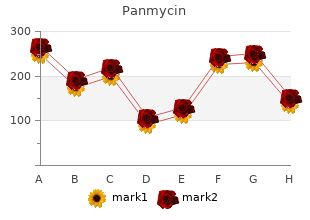
Syndromes
- Infections, including brain abscess, meningitis, encephalitis, and AIDS
- Pelvic prolapse in women -- falling or sliding of the bladder, urethra, or rectum into the vagina, which may be caused by pregnancy and delivery
- Brain tumor or other growth (mass)
- Excessive alcohol use
- Rash
- If your periods have stopped (levels of estradiol vary, depending on the time of month)
- Increased thirst
- You will usually be asked not to drink or eat anything for 6 to 12 hours before the procedure.
With this assumption quality 250 mg panmycin bacterial cell wall, a one-compartment model can be used to predict vancomycin dosage or plasma concentrations (Figure 13-2) discount 250mg panmycin overnight delivery antibiotics for acne minocycline. To predict the appropriate dosage given the desired plasma concentrations buy cheap panmycin 500mg line best antibiotic for gbs uti, we need to know the approximate volume of distribution and the elimination rate constant discount 250mg panmycin fast delivery antibiotics juvenile arthritis. These parameters are used in the multiple-dose infusion equation for steady state also shown in Lesson 5. Because we do not have patient-specific plasma concentration data, the values used for the volume of distribution and elimination rate constant are the population estimates given in the introduction. Note that this equation is similar to those used to determine aminoglycoside dosages: 13-3 (See Equation 5-1. Note that the dose of 1190 mg could have been rounded to 1000 or 1100 mg or rounded up to 1200 mg for ease of dose preparation. The resultant peak from any such dose rounding can easily be calculated with the general equation: (rounded dose)/(actual calculated dose) × (desired peak concentration) In this case, if we rounded our dose down to 1000 mg, the resultant peak calculation is as follows: (1000 mg/1190 mg) × 20 mg/L = 16. The number of doses required to attain steady state can be calculated from the estimated half-life and the dosing interval. To estimate a loading dose, we need to know the volume of distribution and the elimination rate constant. Because we do not know the patient-specific pharmacokinetic values, the population estimates can be used (volume of distribution of 0. Then the equation as shown in Lesson 5 describing plasma concentration over time with an intravenous infusion is applied. Note that again we ignore the distribution phase and assume that a one-compartment model is adequate (Figure 13- 3): (See Equation 13-3. Then, insertion of the known values gives: In this case, the loading dose is not much larger than the maintenance dose. Clinical Correlate Close observation of Figure 13-3 confirms that we are not actually measuring a true peak concentration, as we did for aminoglycosides. We are, rather, measuring a 2-hour postpeak concentration that places this point on the straight-line portion of the terminal elimination phase. After administration of the loading dose (1500 mg) and seven doses (1000 mg each) at 12-hour intervals, plasma vancomycin concentrations are determined to be 29 mg/L (2 hours after the end of the 2-hour infusion) and 15 mg/L at the end of the dosing interval. Although these plasma concentrations are not necessarily harmful, we want to decrease the dosage to attain the original target peak and trough concentrations (20 and 5-10 mg/L, respectively). The information needed to determine a new dosing regimen is the same as described in Problem 1A. However, because we now have data about this specific patient, we no longer have to rely on population estimates. Plasma concentration versus time curve for vancomycin, showing simplification with one-compartment model (dashed line). Calculation of K First, the elimination rate constant (K) is easily calculated from the slope of the plasma drug concentration versus time curve during the elimination phase (Figure 13-4) (see Lesson 3): -1 = 0. Given that the trough concentration will be attained immediately before dose two (given at 8 p. Calculation of elimination rate constant given two plasma concentrations (29 mg/L at 2 hours after the infusion and 15 mg/L at 10 hours after the end of a 2-hour infusion). Calculation of V Note that the elimination rate constant is lower and the half-life greater than originally estimated. Now the volume of distribution (V) can be estimated with the multiple-dose infusion equation for steady state: (See Equation 13-3. These values are then put into the equation: Rearranging gives: So the original estimate for the volume of distribution was close to the volume determined with the plasma concentrations. Calculation of New ττττ Before calculating a new maintenance dose, we can first check to see if we need to use a new dosing interval, as follows: (See Equation 13-4. This every-18-hour dosing interval is a nonstandard interval and can result in administration time errors. Only use intervals such as every 18 or 16 hours when a more standard interval of every 12 or 24 hours does not yield acceptable plasma drug concentrations. Calculation of New K0 Now with the calculated elimination rate constant, volume of distribution, and dosing interval, a revised dosing regimen can be reapplied to solve for the dose. For the concentration at 2 hours after the infusion, we would use the desired concentration of 20 mg/L: Rearranging gives: (See Equation 13-3. In this instance, a peak of more than 20 mg/L is more desirable than a peak less than 20 mg/L, so we would use the dose of 900 mg. The resulting concentration at the end of the dosing interval (trough) can be estimated: (-0. Her recovery is complicated by the onset of acute renal failure 1 week after admission. During the second week, she experiences a spiking fever; gram-positive bacilli, resistant to methicillin but susceptible to vancomycin, are subsequently cultured from her blood. Two hours after the end of a 1000-mg loading dose administered over 1 hour, the vancomycin plasma concentration was 29 mg/L; it is 17. Calculate the vancomycin elimination rate constant, half-life, and volume of distribution in this patient. Note that there are two opportunities to calculate patient-specific pharmacokinetic valuesafter the first dose or after steady state has been achieved. In this case, because the patient has such a long half-life, it is decided to calculate these parameters after the first dose, which allows for subsequent dose adjustments without waiting the many days necessary for steady state to be reached. First, we calculate the elimination rate constant (K) and half-life (T1/2): (See Equation 3-1. Therefore, we must account for the 2 hours that lapsed between the end of the infusion and first plasma level. When calculating the elimination rate constant from two different plasma concentrations, the concentrations should be at least one half-life apart to determine a reasonably accurate slope of the line. Drug concentrations less than one half-life apart can incur great errors in the estimate of the elimination rate constant (K). With the information just determined, calculate when the next vancomycin dose should be given and what it should be. Assume that the plasma vancomycin concentration should decline to 10 mg/L before another dose is given and that the plasma concentration desired 2 hours after the infusion is complete is 20 mg/L. First, we must know the time needed for the plasma concentration to decline to 10 mg/L. It can easily be calculated from the known plasma concentrations, the elimination rate constant, and the desired trough plasma concentration: -Kt Ctrough = Cpeak(steady state)e where: Cpeak(steady state) = observed concentration of 29 mg/L, -1 K = elimination rate constant (0. Next, we determine dosing interval and maintenance dose as follows: (See Equation 13-4. Rearranging to solve for K0: Because 350 mg is used instead of 366 mg, the peak will be 19. Finally, we must check to see what our trough concentration will be after rounding both dose and dosing interval.