

East Carolina University. R. Mezir, MD: "Purchase Betnovate online no RX - Discount Betnovate online".
Such prior information is generated through linkage studies by discount 20gm betnovate with visa acne 4 week old baby, for example order 20gm betnovate amex acne keloidalis nuchae, genotyping distantly related individuals who are both aected by the same condition and dening shared chromosomal regions or by autozygosity mapping in consanguineous families generic betnovate 20 gm with amex skin care procter and gamble. Clinical heterogeneity multiple conditions with similar betnovate 20gm amex acne 5th grade, but not identical, clinical features creates complexity as the conditions are unlikely to be caused by changes in the same gene. Therefore precise phenotypic characterisation is key to successful disease gene iden- tication. Genetic heterogeneity arises where changes in more than one gene can lead to indistinguishable clinical conditions (Table 2. Many common disorders with a genetic basis, including sensorineural deafness, non-syndromal learning disability and retinitis pigmentosa, demonstrate high genetic heterogeneity. Novel gene identication is hindered by the low frequency of mutations among the remaining undiscovered genes. The subsequent, and important, processes for delivery of clinical diagnostic services are substantially hindered by the requirement to screen eectively such a large number of genes. Many rare inherited disorders exhibit more limited heterogeneity, including those dened by our group such as brittle cornea16 and urofacial syndromes. Such alterations result in an individual with tissues with distinct genetic proles. A classic example of this is the dierence between the genetic prole of a tumour compared to surrounding normal tissue. A number of the genes related to the overgrowth disorders have been targets for a number of cancer treatments and therefore immediate exciting therapeutic opportunities have arisen. However, such an approach also identies mutations in introns, regulatory promoters and enhancers or in non-genetic sequences that regulate genes already known to cause rare disorders. The challenges of whole genome analysis, particularly the analysis of larger data sets containing up to 6000 novel sequence variants in each individual and the interpretation of the consequences of the sequence alterations require consideration to determine how this approach will be used to maximally exploit the data produced. There are a number of recognisable approaches that can help to lter such extensive lists of genetic changes: segregation of the putative causal variant with a given phenotype in aected family members and its absence in unaected family members can be helpful. However, for conditions and families where there is only limited family history information this may be impossible, while non- penetrance and variable expression of the phenotype can make interpreta- tion dicult. Comparison of sequences across species and evidence of conservation of amino acid residues indicates a higher likelihood that any change would result in a deleterious eect on the protein. Modelling the potential eects on the resultant protein of an amino acid substitution or the functional eects through disruption of a specic motif can be informative. For a minority of variants, in particular those hypothesised to underlie novel genetic causes of human disease, functional studies using cell culture systems can be employed to examine the eects of specic variants. Such approaches can be further complemented by animal models, including in Drosophila, zebrash and mice with dened genetic alterations. Currently most functional and/or animal studies do not have the throughput to be practical to inform routine diagnosis, but where available are useful in providing evidence to support the role of the causative gene. The majority of these tests are still undertaken on a research basis in a range of laboratories. The traditional testing model has been for a clinician to dene, through detailed clinical investigation, a specic phenotype and to develop a clinical hypothesis. This would result in the ordering of a specic genetic test on a single gene (or at most a very small number of potentially relevant genes) to test that hypothesis. The pick-up rate of such a testing approach varies considerably, from approximately 0. In general this has been an inecient approach which is by its very nature limited to patients, and their relatives, with phenotypes consistent with a genetic disease. Testing has been espe- cially challenging in heterogeneous conditions, including developmental View Online Diagnosis of Rare Inherited Diseases 45 delay, deafness, retinal dystrophies and glycogen storage disorders. The development of panel testing, where a selected array of genes can be analysed in a single assay, has been successfully introduced. Our own experience with testing of a panel of 105 retinal dystrophy genes has seen an increase in detection of the causal variant from 14 to 60% over the past 2 years of providing this service. At present clinical reports are generated providing feedback on specic phenotypes relevant to the presentation of the tested individual. Reports may also provide information about carrier status for a range of recessive disorders, so informing future reproductive risks, and of unexpected dominant disorders for which preventive screening may be appropriate. Initial clinical exome testing has focused on the testing of children with learning disabilities, developmental disorders and neurological phenotypes. Studies have assessed the utility of exome testing in a number of settings including improving diagnosis of children on intensive care units or aected by likely recessive disorders when born to consanguineous parents. The next chal- lenge is to introduce this testing into other areas of mainstream medicine including cardiology, renal and gastrointestinal medicine. A number of studies have started to consider how this extra information generated from exome or genome analysis should be fed back to tested individuals. Information about increased risks of coronary artery disease, cancer and rare inherited disorders like Marfan syndrome lend themselves to targeted interventions. However, concerns have been raised about individual autonomy, inappropriate use of this information to discriminate in terms of employment and insurance and the burden placed upon health profes- sionals to feed back accurate information that can have a benet rather than indicating increased risk with no potential to alter natural history, for example in providing information about neurodegenerative disorders. The improved technology, reduction in costs and advances in bioinformatics mean that exome sequencing and in time whole genome sequencing will become routine in clinical diagnosis over the next decade. Many challenges exist to ensure that the potential is harnessed to improve health care but the opportunities are too great for this not to happen. Identication of the genetic cause of individual disorders through exome sequencing oers enormous opportunity for personalised medicine. Gene therapy based approaches based on knowledge of the specic altered gene or alternative strategies, e. Already examples have emerged of individuals who have had successful diagnosis and treatment due to exome sequencing. These criteria, a prevalence of <200 000 and an incidence no greater than 5 in 10 000 persons respectively, provide acceptable operational denitions for what constitutes a rare disease. There are also a number of regulatory enablers that can help to facilitate clinical development of drugs for rare diseases. By their nature, clinical trials for rare diseases are conducted in small patient populations. However drugs developed for rare disease populations are subject to the same rigour in the assessment of safety and ecacy as drugs developed for more common diseases. While the number of clin- ical trials described in the label was smaller for orphan than for non-orphan drugs (2. In rare diseases with small target patient populations, incomplete understanding of the disease process, lack of validated measures for disease activity, incomplete understanding of the standard of care and a small target population from which to recruit may present unique challenges that must be mastered if the study hypothesis is to be properly tested. These challenges may include (i) incomplete understanding of the disease clinical course, with a resulting adverse impact on ability to anticipate outcomes for placebo or active comparator treatment groups; (ii) lack of validated measures of disease activity/progression resulting in limitations on end point(s) capable of supporting regulatory approval in a reasonable time frame; (iii) incomplete understanding of the standard of care for disease management, potentially increasing the heterogeneity that may be encountered among subjects in small data sets; (iv) small numbers of potential subjects, which creates diculty accessing sucient sample size to support hypothesis testing and/or characterisation of benet risk. Engaging a sucient number of study sites to support subject recruitment plus heightened competition with other sponsors/investigators for the limited quantum of patients available for investigation may create logistical challenges. For disorders of genetic origin, this oen results in aggregation of groups of (i) individuals aected by the same mutation against a background of diering background genetic modiers, (ii) individuals aected by dierent mutations in the same gene, or (iii) by mutations in dierent genes that contribute to a common physiologic pathway. For acquired disorders, individuals with similar path- ologic outcomes are oen aggregated despite incompletely understood dierences in underlying aetiology. The resulting limitations in clinical experience and poorly understood heterogeneities in patient populations contribute to an incomplete understanding of the disease clinical course and adversely impact the ability to anticipate outcomes for placebo or active comparator treatment groups.
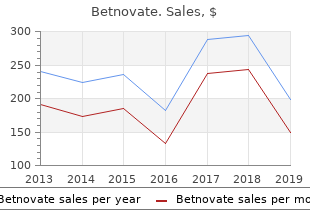
Diseases
- Growth retardation hydrocephaly lung hypoplasia
- Chromosome 2, trisomy 2q
- Decompression sickness
- Yemenite deaf-blind hypopigmentation syndrome
- Lichen sclerosus et atrophicus
- Osteogenesis imperfecta congenital joint contractures
- Ahumada-Del Castillo syndrome
As the disease progresses buy cheap betnovate 20gm online b5, the patient may note gait difculties order betnovate 20 gm with visa acne nodule, micrographia (shrinkage in size of handwriting) cheap betnovate 20 gm with amex skin care 7, and issues with axial posture [9] order betnovate 20 gm overnight delivery acne natural treatment. Other changes related to motor function include decreased facial expression and laryngeal dysfunction resulting in difculties with speech [10 ]. These include the serotonergic, cholinergic, and noradrenergic networks, effects on which may explain some of the non-motor symptoms associated with the disease [12 15]. Cause of death include pulmonary infections due to aspiration of saliva into the lungs, accidents associated with increased risks of falls due to loss in motor function, chocking due to difculty swallowing, and blockage of blood vessels leading to pul- monary embolism and deep vein thrombosis. Progression of symptoms is also associated with the presence of abnormal intra- neuronal proteinacious accumulations called Lewy bodies that occur in both neuro- nal cell bodies and in neurites [6, 23]. Extent and expression of the disease at dened sites within the nervous system has been proposed to track closely with the presence of Lewy bodies at these various locations and may in part explain patient-to-patient variabil- ity in symptoms [19]. If correct, this could have major implications in terms of the success of proposed therapies for the disorder, including use of cellular transplantation (see below). However there are clearly caveats to the data supporting this theory that are under intense investigation [29]. Existing therapies are instead primarily directed towards alleviating the most disturbing symptoms in individuals with the disease, which can vary widely in terms of both presence of particular symptoms and rate of progression [7]. For this reason, there is no standard treatment for the disorder and treatments must be tailored to meet the needs of each individual patient. It is particularly effective in reducing bra- dykinesia and rigidity in early stages of the disease. It is often given in conjunction with carbidopa, a peripheral dopa decarboxylase inhibitor that acts to lessen side effects due to conversion of L-dopa to dopamine outside of the brain, which can lead to nausea and vomiting [31]. These side effects are controlled in many cases by addition of other drugs to the patient s medication regime [32]. These include Requip, Mirapex, and Neupro that are taken either alone or in combination with Sinemet (L-dopa/carbidopa) [36]. They however have their own side effects, including increased risk for drug-associated psychosis [37]. Sometimes there are additionally dietary considerations due to interactions of medications with specic foods [38]. Other recommended evidence-based treatment approaches include general life- style modications such as rest and exercise for improvement of motor function and reducing stress, and speech therapy to aid with losses in communication abilities [44 46]. Other over-the-counter nutriceuticals (low-dose lithium, vitamin D, nicotine, etc. Over the last decade, there has been a large amount of research put into novel restorative therapies including gene therapy and cellular replacement via transplan- tation. However, results from various clinical trials proved to be contradictory, perhaps due to differences in trial design. Other confounding factors include hetero- geneity of the patient population and the difculties inherent in teasing out symp- tomatic versus drug-mediated effects. As with gene therapy, several clinical trials using cellular transplantation have been undertaken, as early as the 1980 s. Study results have been variable, but more detailed placebo-controlled double-blind trials (versus open-labeled) gener- ally report lack of signicant overall improvement in motor function and the induc- tion of drug-related side effects such as dyskinesia. However, excitement has been somewhat tempered based on evidence suggesting that transplanted cells have limited success in clinical trials and may actually take on the same fate as affected endogenous neurons [28]. More studies are undoubtedly required before moving such studies towards new clinical trials, including how to deal with reduced transplant efciency in the older brain. In other words, not only do we need to consider the cells themselves, but also the envi- ronment into which they are placed. These vary from individual to individual, which may to some degree help explain disparate disease presentation. Advanced age is certainly directly linked to a more rapid disease progression and older indi- viduals are more refractory to medical treatments for the disorder, suggesting that there is an important interplay between the two [67 ]. Identication of these molecular targets has led to exploration of interven- tions designed to prevent or reverse their detrimental effects as a means of slowing or reversing the course of the disease. These have been helpful in the segmental dissection of different aspects of disease pathology, including the role of mitochondrial defects in neuropathological features associated with the disease [73 ]. However, use of agents that act to increase mitochondrial biogenesis will need to be balanced with those which increase lysosomal turnover of defective mitochondria so as to not increase the build-up of the latter. Enhancement of ssion-fusion events in early stages of the disease may also be effective in repairing damaged mitochondria. However, as levels of damaged mitochondria increase, these pro- cesses lose their effectiveness and are replaced by removal of dysfunctional mito- chondrial via lysosomal degradation. A recent clinical trial using mitochondrially targeted CoQ10 (MitoQ) also failed to demonstrate slowing of clinical progression of the disease [83]. Losses in mito- chondrial function can affect the ability of the organelle to sequester calcium and this in turn can result in the generation of mitochondrial-mediated oxidative stress and subsequent damage to the organelle that can further affect its function [85 ]. In addition, preclinical studies in various animal models strongly suggest the involvement of inflammatory processes in associated neuronal cell death [95]. Neuroinflammatory processes may contribute to deleterious events lead- ing to neuronal degeneration. Possible factors involved in neuroprotection may include inhibition of cyclooxygenase 2 and reduced production of prostaglandins [100, 101]. Neuroinflammatory processes represent an attractive therapeutic target for slowing progression of the disorder. During aging, astrocyte numbers increase and a greater proportion become acti- vated (astrogliosis) [104, 105]. This is another potential target for therapy, but although prion-like disease spread is known to require endo- somal release and uptake, much more about the molecular mechanisms involved in this process still needs to be elucidated. There is however some concern that this may prevent potentially neuroprotective aggregation of the protein, and this may actually exac- erbate the condition. It is also conceivable that post-translational modications that drive aggregate formation may evade antibody binding and may increase neuroin- ammation (see above). Unfortunately, attempts to intervene have to date provided little to no benet in human clinical trials [51]. It may also reect lack of thoroughly vetted pre-clinical studies in order to better understand dose responses, treatment pharmacokinetics, and appropriate therapeutic windows. Identication of a disease-modifying neuroprotective intervention has remained elusive to date, likely due in part to these factors. It may however also imply that exploration of novel targets far aeld from those conventionally studied is needed. While this has yielded important information and enormous efforts have been expended to develop therapies based on these ndings, there have been few successful interventions as a consequence. This is likely due to an incomplete picture of the nature of complex chronic disease states. Studying the role of aging mechanisms across a wide variety of disease states will allow scientists to broaden the scope of research beyond tradi- tional disciplines, towards the central concept that these multiple human disease states likely arise from a common underlying cause: aging itself. The term gerosci- ence was coined by scientists at the Buck Institute for Research on Aging in 2007 as an acknowledgement and organizing principle of this scientic concept.
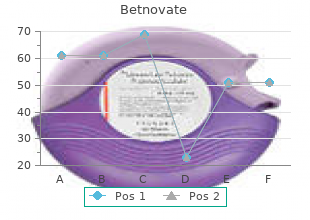
Diseases
- Blepharoptosis myopia ectopia lentis
- Chromosome 7, monosomy
- KID syndrome
- Chromosome 11, deletion 11p
- Gemignani syndrome
- Chromosome 3, monosomy 3p25
- Mansonelliasis
- Cleft lip palate dysmorphism Kumar type
- Cholecystitis
- Spondylarthropathy
Leishmaniasis should always be considered in returning travelers discount betnovate 20gm line skin care 1 month before marriage, but diphtheria is probably often overlooked [4 order betnovate 20 gm fast delivery skin care 11 year olds,5] cheap betnovate 20 gm without prescription acne aid soap. However generic 20gm betnovate overnight delivery acne zones, there are only few published studies available on the prevalence or the incidence of pyoderma under tropical conditions [6]. A study performed in Blantyre, Malawi did not show a high incidence of ulcerating pyoderma at the in- and the out- patient population at a hospital [7]. Skin lesions and the upper respiratory tract are the primary focal sites of infection. It seems that at least a minor trauma is necessary for the devel- opment of streptococcal pyoderma. Since protecting clothing is used less under tropical conditions, minor trauma of the skin is more likely to occur providing a port of entry for an infection. Carrier sites are the anterior nares, the perineum, the axillae, and the toe webs. Infec- tion may be initiated after colonization of skin lesions, especially moist Ulcerating Pyodermas 109 lesions. Whether an infection is contained or spreads depends on sev- eral complex factors such as the host defense mechanisms and the viru- lence of the S. Several toxins and enzymes such as protease, lipase, and hyaluronidase contribute in the invasion and the destruction of tis- sues. However, systematic surveillance data on the prevalence from trop- ical countries are limited. Some data are available on antimicrobial resis- tance, which shows methicillin-resistant strains as an increasing problem. Special attention must be paid to the community-acquired methicillin- resistant S. It has recently been reported as a cause of complicated soft tissue infections in travelers returning from nontemperate climates [9]. Cutaneous diphtheria is still endemic in tropical countries, whereas it is generally travel-related and currently rare in the developed world because of the policy of routine active immunization. Recent large epidemics in eastern Europe have again drawn attention to this disease. Cutaneous diphtheria may be encountered more often in the near future because of the increased travel to and from the endemic countries. Ecthyma is known as a clinical entity and is characterized by a deep pyogenic ulcerating infection. It usually starts as a vesicle or vesiculopustule on an erythematous base, which subsequently ulcerates. There are usually few lesions, but new lesions may develop without adequate treatment. Pyoderma in travelers is generally encountered as a secondary infection in the skin lesions caused by environmental insults such as insect bites, abrasions, scabies, and atopic dermatitis to the skin. Systemic toxic complications such as myocarditis and neuritis are rare in immunized individuals. The most typi- cal manifestation is characterized by a chronic, nonhealing ulcer(s) with a punched-out appearance, slightly undermined and covered with a gray adherent membrane. It is painful in the rst 2 weeks, becoming painless later, and the hemorrhagic base appears after the (spontaneous) removal of the adhering crust. This is often not rou- tinely performed in daily practice because it is more time-consuming and inconvenient for the patient. The ulcerated lesion should be thoroughly cleansed with saline solution after which specimens from the wound surface and if possible, from under the margins of the wounds should be collected. The denite diagnosis is established after isolat- ing and identifying the organism from the ulcer and demonstrating its toxigenicity. Treatment Treatment of ulcerative pyoderma is initially based on the clinical assess- ment. Gram-stained smears of exudate may be helpful in starting empirical antimicrobial treatment. When culture results are not available, but the clinical condition demands antibiotic treatment, one may start with ucloxacillin orally for at least 10 days as drug of rst choice in cases of community-acquired ulcerative pyoderma. The antibacterial activity of ucloxacillin is evident on Gram- positive bacteria and above all in penicillinase-producing staphylococci. Macrolide antibiotics should be prescribed with caution considering the global spread of macrolide-resistant S. Although there is evidence that topical antibiotic treatment is effective, in extensive pyoderma topical antibiotic treatment is not recommended. It is generally accepted that ulcers heal more rapidly under occlusive (moist) dressings. There are no documented studies in which the effect Ulcerating Pyodermas 113 of these occlusive dressing on healing of ulcerative pyoderma was shown. However, occlusive dressings were shown to be safe in chronic ulcers with an even lower infection rate in occluded wounds compared with ulcers treated with conventional dressings. Special attention should be paid to ulceration in the lower legs in which (sub)clinical edema is often present. Edema may be eliminated by adequate compression therapy with elastic or nonelastic bandages or stockings. Treatment should be started as soon as possible when cutaneous diph- theria is suspected. Penicillin and erythromycin are considered drugs of rst choice for eradicating C. The family Rickettsiaceae consists of two genera, the genus Rickettsia and the genus Orientia. The genus Rickettsia is divided into two groups based on differ- ences in lipopolysaccharides, outer membrane protein A, and evolutionary genetic relationships: the typhus group and the spotted fever group [1 3]. Rickettsioses occur all over the globe and are increasingly recognized in travel medicine [1, 4 6]. Increased expo- sure in endemic areas due to adventure (eco)tourism [4] and military operations [7] also plays a role in the reported increase of cases. Vari- ous mammals play a role as reservoir but ticks, vector for many Rickettsia species, are also important as reservoir because of transovarial transmis- sion. Transovarial transmission is less important in eas and mites and does not occur in lice. They can be cultured in eggs and in chick embryo cells and various mammalian and arthropod cell lines [8]. Culturing is only per- formed in specialized laboratories under strict safety conditions. A rash appears about 3 5 days after onset, often rst macular evolving to maculopapular.